Chaque être humain possède plus de 20 000 protéines. Par exemple l’hémoglobine qui s’occupe du transport de l’oxygène depuis les poumons vers les cellules de tout le corps, ou encore l’insuline qui indique à l’organisme la présence de sucre dans le sang.
Chaque protéine est formée d’une suite d’acides aminés, dont la séquence détermine son repliement et sa structure spatiale – un peu comme si un mot se repliait dans l’espace en fonction des enchaînements de lettres dont il est composé. Cette séquence et ce repliement (ou structure) de la protéine déterminent sa fonction biologique : leur étude est le domaine de la « biologie structurale ». Elle s’appuie sur différentes méthodes expérimentales complémentaires, qui ont permis des avancées considérables dans notre compréhension du monde du vivant ces dernières décennies, et permet notamment la conception de nouveaux médicaments.
Depuis les années 1970, on cherche à connaître les structures de protéines à partir de la seule connaissance de la séquence d’acides aminés (on dit « ab initio »). Ce n’est que très récemment, en 2020, que ceci est devenu possible de manière quasi systématique, avec l’essor de l’intelligence artificielle et en particulier d’AlphaFold, un système d’IA développé par une entreprise appartenant à Google.
Read more:
L’intelligence artificielle au défi du design de protéines : des prouesses et limites d’AlphaFold
Face à ces progrès de l’intelligence artificielle, quel est désormais le rôle des chercheurs en biologie structurale ?
Pour le comprendre, il faut savoir qu’un des défis de la biologie de demain est la « biologie intégrative », qui a pour objectif de comprendre les processus biologiques au niveau moléculaire dans leurs contextes à l’échelle de la cellule. Vu la complexité des processus biologiques, une approche pluridisciplinaire est indispensable. Elle s’appuie sur les techniques expérimentales, qui restent incontournables pour l’étude de la structure des protéines, leur dynamique et leurs interactions. De plus, chacune des techniques expérimentales peut bénéficier à sa manière des prédictions théoriques d’AlphaFold.
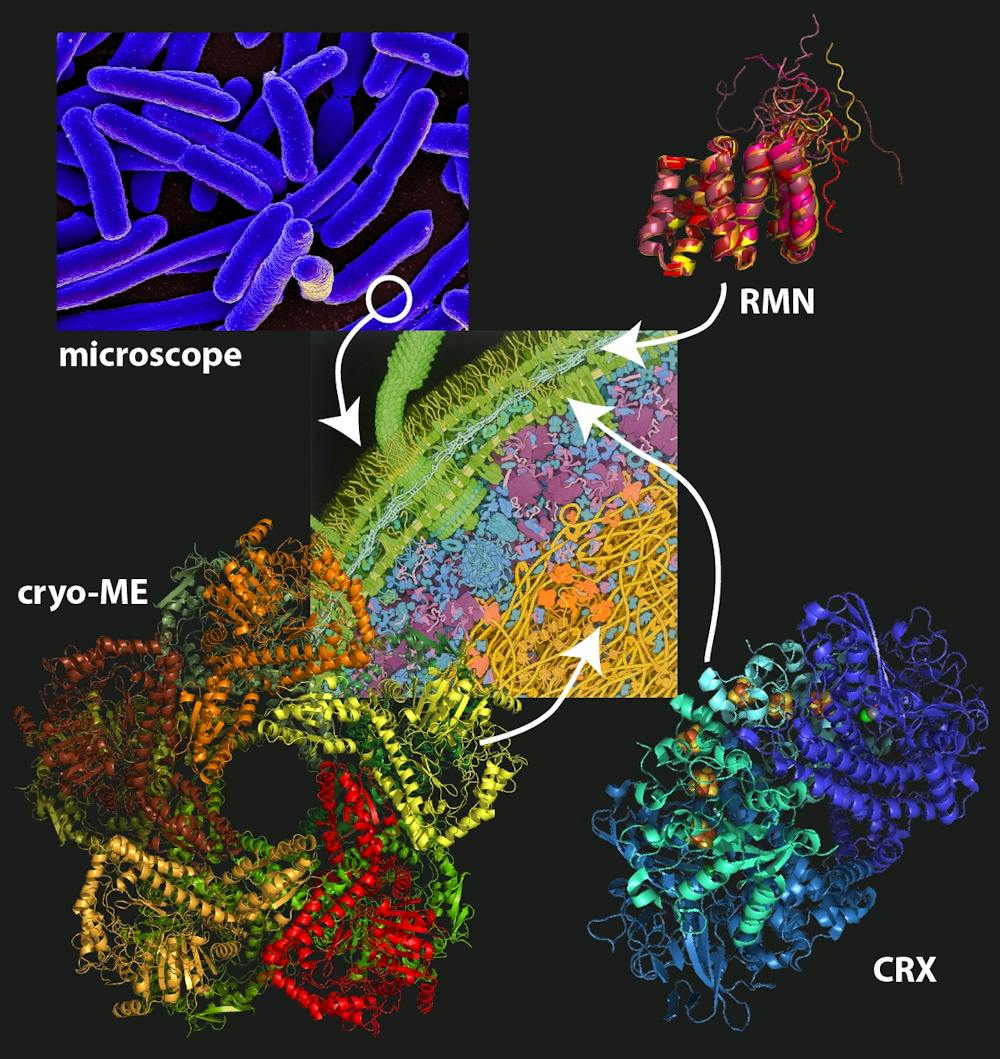
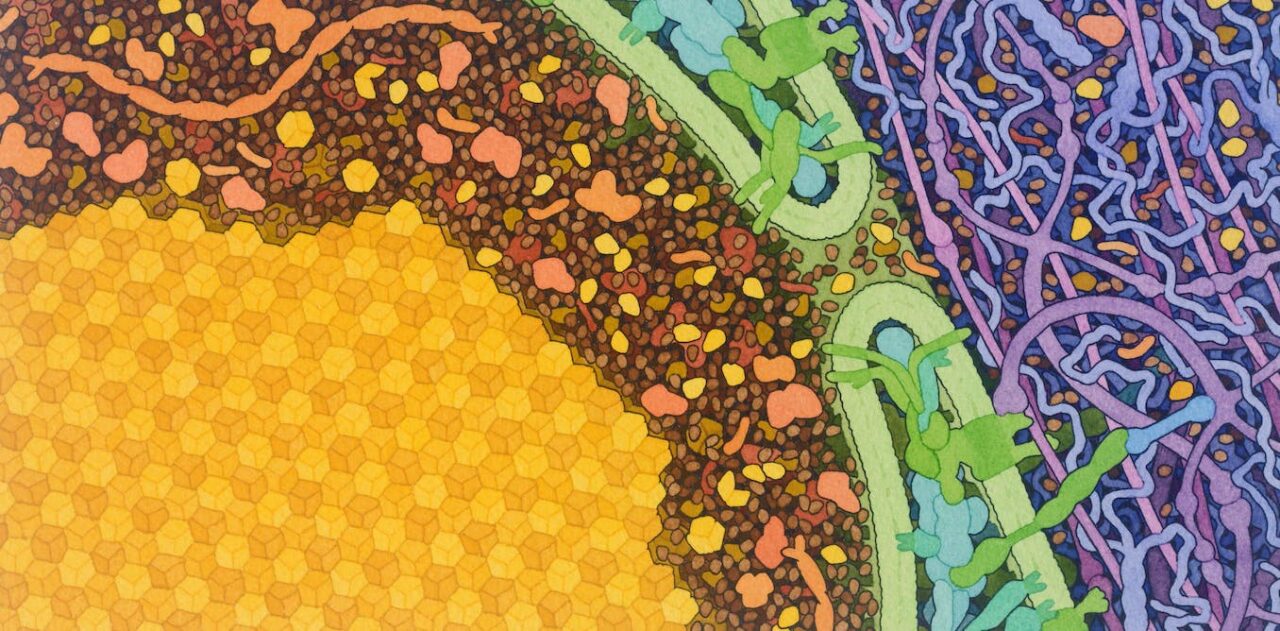
Les structures de trois protéines de la bactérie Escherichia coli, déterminées par les trois méthodes expérimentales expliquées dans l’article, à l’Institut de Biologie Structurale de Grenoble.
Beate Bersch, IBS, à partir d’une illustration de David Goodsell, Fourni par l’auteur
La cristallographie aux rayons X
La cristallographie est, à cette date, la technique la plus utilisée en biologie structurale. Elle a permis de recenser plus de 170 000 structures de protéines dans la « Protein Data Bank », avec plus de 10 000 repliements différents.
[Près de 80 000 lecteurs font confiance à la newsletter de The Conversation pour mieux comprendre les grands enjeux du monde. Abonnez-vous aujourd’hui]
Pour utiliser la cristallographie à rayons X, il faut faire « cristalliser les protéines ». On dit souvent que cette technique est limitée par la qualité de cristaux de protéines, qui est moindre pour les grosses protéines. Mais cette notion ne correspond pas toujours à la réalité : par exemple, la structure du ribosome, l’énorme machine moléculaire qui assemble les protéines, a été résolue à 2,8 angströms de résolution. Venkatraman Ramakrishnan, Thomas Steitz et Ada Yonath ont reçu le prix Nobel de chimie en 2009 pour ce travail.
Avec le développement récent du laser X à électron libre (XFEL), il est devenu possible d’étudier simultanément des milliers de microcristaux de protéines à température ambiante et à l’échelle de la femtoseconde (10-15 secondes, soit un millionième de milliardième de seconde, l’échelle de temps à laquelle ont lieu les réactions chimiques et le repliement des protéines). Cette technique permet d’imager les protéines avant qu’elles ne soient détruites. Elle est en train de révolutionner la « cristallographie cinétique », qui permet de voir les protéines « en action », ainsi que la recherche de médicaments.
Pour l’instant, l’apport d’AlphaFold à l’étude de la structure des protéines par cristallographie s’est concentré dans la génération de…
La suite est à lire sur: theconversation.com
Auteur: Beate Bersch, Chercheuse CNRS à l’Institut de Biologie Structurale, Université Grenoble Alpes (UGA)
